Equipe du Dr Pascal Barbry
Physiologie génomique des eucaryotes | Public
expert | Grand
public |
Chaire 3AI de biologie computationnelle
 |
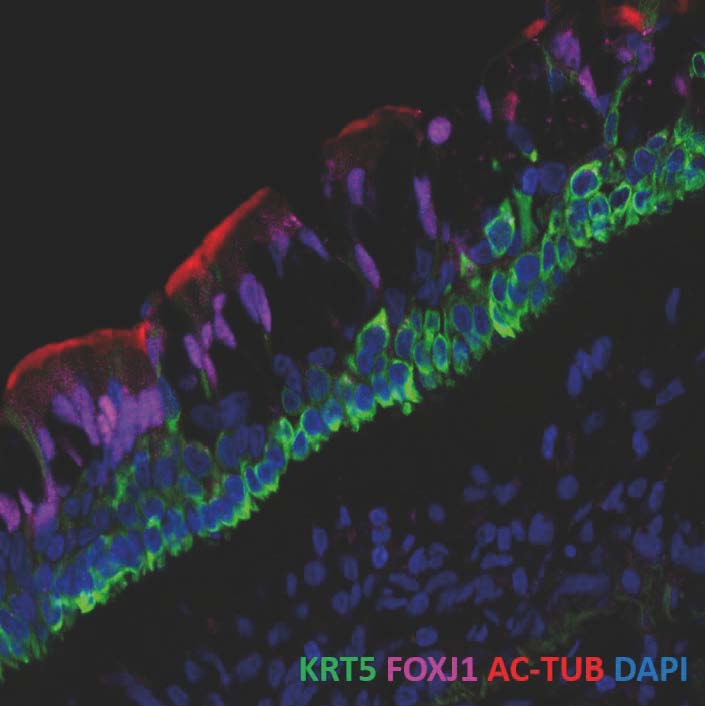 L’épithélium des voies respiratoires : un écosystème protecteur
L’épithélium des voies respiratoires : un écosystème protecteurL’épithélium des voies respiratoires tapisse le nez, la trachée et les bronches. Il est constitué de différents types de cellules qui orchestrent la clairance mucociliaire afin de protéger efficacement la muqueuse des voies respiratoires contre différents stress (allergènes, composés chimiques, bactéries, virus, transformation, etc.). Ces types cellulaires sont produits à travers de programmes génétiques spécifiques, qui aboutissent à la différenciation de cellules épithéliales multiciliées, présentant à leur surface apicale des centaines de cils motiles, de cellules à mucus, capables de secréter un mucus protecteur qui tapisse les surfaces épithéliales et de différents types de cellules basales qui jouent un rôle dans l’ancrage et la régénération de l’épithélium. Tout dysfonctionnement qui les affecte peut être associé à une maladie grave comme l’asthme, la bronchopneumopathie chronique obstructive (BPCO) ou la mucoviscidose.
Notre équipe s’intéresse aux programmes génétiques de différenciation qui permettent la construction de l’épithélium des voies respiratoires. Pour les étudier, nous disposons de plusieurs modèles de cultures primaires de cellules humaines, capables de reproduire certaines conditions physiologiques essentielles.
Nous avions découvert en 2011 le rôle déterminant joué par les familles de micro ARN (miRNA) miR-34/449, dans la différenciation des cellules multiciliées (processus appelé multiciliogénèse), en bloquant le cycle cellulaire, la voie Notch, puis la relocalisation du cytosquelette d’actine. De façon surprenante, plusieurs autres molécules (CCNO, MCIDAS, CDC20B) également codées par la même région génomique 5q11 chez l’homme, ont toutes été impliquées dans les mécanismes de multiciliogenèse. C’est notamment le cas de CDC20B qui participe au fonctionnement du deutérosome, organelle cytoplasmique spécialisée qui permet la multiplication des centrioles, pré-requis indispensable à la biosynthèse des multiples cils motiles des cellules multiciliées chez les vertébrés. Nos études s’intéressent également aux mécanismes moléculaires contrôlant l’expression des cellules à mucus.
L’étude du transcriptome à l’échelle de la cellule uniqueDepuis le début de l’année 2015, nos recherches sur l’épithélium des voies aériennes se sont dotées d’une nouvelle approche permettant une étude à l’échelle de la cellule unique, dans le cadre d’un projet soutenu par la Cancéropôle PACA, l’association Vaincre la Mucoviscidose, l’infrastructure nationale France Génomique et le Conseil Départemental des Alpes Maritimes. Les travaux menés sur plusieurs technologies de séquençage en « single cell » (C1 Fluidigm et Chromium 10X Genomics) ont permis d’acquérir des informations sur plusieurs milliers de cellules dont les propriétés sont désormais étudiées en détail par notre équipe.
 | 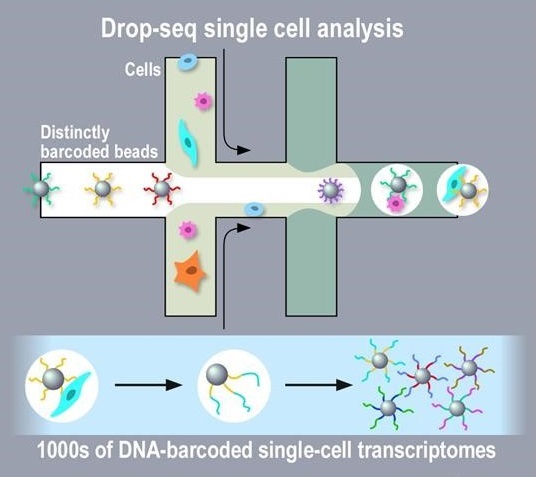 |
| C1 Fluidigm | Chromium 10X Genomics |
Grâce aux données ainsi générées, nous étudions non seulement le développement tissulaire normal, tel qu’il se déroule au cours du développement, mais aussi dans des conditions reproduisant des conditions pathologiques, dans lesquelles des modifications des profils de différenciation sont causées par des niveaux importants de certaines cytokines. Nous travaillons actuellement à modéliser de la façon la plus parcimonieuse les réseaux de régulation génique mis en jeu.

Le
« Human Cell Atlas » est un effort international visant à créer un atlas de référence de toutes les cellules du corps humain. Il doit permettre d’établir une ressource accessible à tous pour des études en biologie/médecine. Notre équipe est la première équipe française à y avoir été associée. Elle y contribue à travers la construction de l’atlas des poumons et des voies respiratoires, qui ont d’ailleurs permis d’identifier les cellules pouvant fixer le virus du SARS-CoV-2 au niveau des voies aériennes. Ces travaux ont été publiés dans plusieurs journaux depuis 2019 (American Journal of Respiratory & Critical Care Medicine, Nature Medicine, Development).
Lorsque nous considérons la quantité énorme de cellules présentes dans le corps d'un homme (~10
13 cellules) ou même seulement d'une souris (~10
10 cellules), il est difficile d’appréhender la diversité des types cellulaires présents, tous pourtant issus du même génome : cellules nerveuses, cellules musculaires, cellules immunitaires… Chaque type cellulaire diffère des autres par l’expression de certains gènes, l’apparition de variants d’épissage, etc. Acquérir suffisamment d’information sur les différents gènes exprimés dans une cellule peut permettre de l’identifier, mais aussi révéler son fonctionnement ou un état particulier. Une telle information peut être exploitée pour mieux comprendre des étapes clés du développement embryonnaire, ou le déclenchement de certaines maladies. Plusieurs projets internationaux réalisent actuellement des atlas. Ainsi, le « Human Cell Atlas » cherche à cataloguer les types et états cellulaires existants chez l’homme à partir de mesures de l'expression des gènes à la résolution d’une cellule unique. De tels atlas vont bientôt représenter des systèmes de positionnements globaux que les biologistes pourront utiliser pour répondre à toutes sortes de questions : Combien y a-t-il de types cellulaires dans un organe ? Qu’est-ce qui différencie des types cellulaires identiques d’organes différents ? Comment des cellules voisines communiquent-elles ?
C’est ainsi que le consortium Human Lung Biological Network a réussi à caractériser les portes d’entrée du virus du SARS-CoV-2 à l’intérieur de l’organisme.
Plus d'informations...
Lire l’ARN de tout son long, cellule par cellule
Nous avons décrit en 2020, dans un article publié dans Nature Communications (https://pubmed.ncbi.nlm.nih.gov/32788667/), une nouvelle méthode pour analyser plus finement la structure des ARN, afin d’y détecter des variations d’épissage ou d’édition avec une résolution allant jusqu’à la cellule unique. Ces approches combinent des outils de microfluidique avec le séquençage des acides nucléiques sur de grandes longueurs. Leur développement vise à améliorer la construction des atlas de cellules uniques qui sont en cours de réalisation.
De nombreux protocoles sont déjà disponibles pour analyser à la résolution de la cellule unique les mutations somatiques ou la méthylation de l'ADN, l'accessibilité de la chromatine, les profils épigénétiques, ou les variations du nombre de copies. L'approche transcriptomique sur cellule unique (scRNA-seq) reste pour l’heure l’approche la plus globale, permettant une analyse qualitative du transcriptome sur plusieurs centaines de milliers de cellules.
Une limite actuelle du scRNA-seq réside dans la façon dont s’effectue le séquençage, qui se limite le plus souvent à une seule extrémité de chaque molécule d’ARN. C’est suffisant pour décrire quantitativement le niveau d’expression des gènes, mais les informations sur l'épissage et l'hétérogénéité des séquences, réparties sur l’ensemble du transcrit, sont le plus souvent perdues.
Pour obtenir des informations plus complètes sur la séquence du transcriptome, nous avons mis en place une approche de séquençage pleine longueur à l’aide d’un séquenceur Nanopore.
La méthode mise au point a été baptisée ScNaUmi-seq. Le ScNaUmi-seq pourrait ainsi permettre d'analyser le paysage mutationnel des tumeurs, ou encore documenter les mécanismes d’épissage mis en place lors du développement précoce (https://pubmed.ncbi.nlm.nih.gov/34497388/). Ce développement ajoute donc une nouvelle couche d'information importante pour améliorer notre définition des différents types cellulaires.
Un article plus récent (https://www.biorxiv.org/content/10.1101/2020.08.24.252296v1), actuellement accessible sous forme de pré-publication, prolonge ce travail en appliquant la même approche à des échantillons de transcriptomique spatiale. La méthode, baptisée Spatial isoform Transcriptomics (SiT) permet de définir des variations spatiales d’épissage et/ou d’édition du génome.
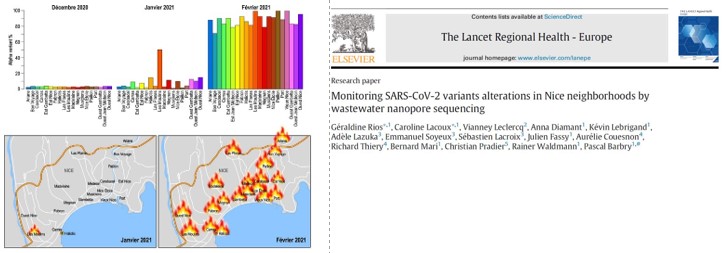
Nous avons rapporté en août 2021 dans la revue Lancet Regional Health Europe comment la dissémination du variant « anglais » (alpha) du SARS-CoV-2 s’est opérée à l’intérieur de la ville de Nice au début de l’année 2021. Nous avons obtenu ces résultats en analysant les eaux usées d’une vingtaine de points de la ville par séquençage d’ARN. Cela nous a permis de dresser une carte des différents variants présents sur le territoire. De telles approches fourniront certainement dans le futur de précieuses informations épidémiologiques sur ce virus ou tout autre matériel infectieux.
Le génome du virus SARS-CoV-2 contient environ 30000 ribonucléotides. Nous avons mis en place une approche de séquençage par nanopores pour suivre les différents variants du virus du SARS-CoV-2 dans les eaux usées de la ville de Nice qui permet de quantifier la concentration du virus tout en déterminant également la nature des éventuels variants. Nous avons démontré que les quantifications effectuées dans les eaux usées de différents quartiers d’une ville permettaient de bien refléter ce qui se passait en surface, après avoir comparé les signaux mesurés dans les eaux usés aux mesures effectuées sur des échantillons de patients récupérés durant la même semaine. Des mesures effectuées sur les réseaux d’assainissement donnent ainsi une image assez précise de ce qui peut se passer à l’échelle de toute la population, en permettant le criblage des personnes symptomatiques aussi bien que des personnes asymptomatiques.
Avec l’aide des services de l’assainissement de la Métropole de Nice, des eaux usées provenant de 20 quartiers différents de la ville ont été collectées. Des informations très précises sur les différents variants qui circulaient ont alors pu être collectées pendant 6 mois, permettant, d’alerter très tôt en 2021 les autorités sur la présence de variants « à risque » dans certains quartiers de la ville.
Nos mesures ont permis de détecter dès janvier 2021 l’émergence dans un des quartiers de la ville d’un variant particulier de la lignée alpha, porteur d’une mutation A522S sur la spicule. Ce variant n’avait été détecté que chez moins de 2% de tous les virus B.1.1.7, mais c’est pourtant cette souche qui s’est rapidement répandue dans toute la ville pour y devenir largement majoritaire à partir du mois de février.
Stockage de données : les promesses de l’ADN synthétique
Le projet e uropéen OligoArchive travaille à établir des preuves de concept pour un stockage de données sous la forme de collection de courtes séquences d’ADN synthétique. L’ADN représente un support en théorie inégalé en termes de densité d’information et de longévité. Le projet vise à lever certaines limitations techniques qui restent à surmonter, largement liées au coût important de production des séquences.
uropéen OligoArchive travaille à établir des preuves de concept pour un stockage de données sous la forme de collection de courtes séquences d’ADN synthétique. L’ADN représente un support en théorie inégalé en termes de densité d’information et de longévité. Le projet vise à lever certaines limitations techniques qui restent à surmonter, largement liées au coût important de production des séquences.
Ce travail fait écho à l’explosion des données liées au développement d’internet et de la téléphonie mobile. La quantité totale de données stockées devrait représenter deux millions de milliards de milliards de bits d’ici à 2025. Toutes ces données tiendraient dans l’équivalent de vingt-trois piles de disques Blu-ray allant de la surface de la Terre jusqu’à la Lune. Cette situation force les géants d’Internet à multiplier les data centers, de plus en plus souvent implantés dans des zones froides à cause de leurs besoins gigantesques en refroidissement. Dans la quête de systèmes de stockage mieux adaptés, l’ADN dispose d’avantages intéressants : un seul gramme d’ADN peut contenir jusqu’à 455 exabits d’informations, soit 455 milliards de milliards de bits. Toutes les données du monde tiendraient alors dans une boîte à chaussures. Son stockage à l’abri de l’oxygène et de l’eau permet une conservation pendant plusieurs milliers d’années à température ambiante, ce qui en fait un moyen de stockage plus stable que le stockage numérique sur bandes.
Le challenge relevé par le projet européen OligoArchive consiste à créer des séquences d’ADN capables de contenir, en moyenne, encore plus de données numériques pour un même nombre de nucléotides, ce qui permettra de réduire les coûts de synthèse, de corriger automatiquement les erreurs lors du séquençage, et de mettre en place des moyens de lecture et de relecture simples de l’information.
 |
| Images numériques après codage et synthèse sur ADN. À gauche, séquençage et décodage au moyen d’une solution de compression non adaptée ; à droite, séquençage et décodage au moyen de la solution de compression développée par le projet OligoArchive. |
Photo de l'équipe